If you’ve come to this post excited in the hopes of creating your own superhuman buddy, I’m sorry to disappoint (I really am because that sounds awesome!). Sadly, I’m not making mutants in the sense of Wolverine but mutating a culture of cells to contain a specific gene. As mentioned in a previous post, I’m currently working on my Master’s project investigating the effects of phosphorylation on a protein called DEF6. By mutating certain serine and threonine amino acids which are known to be phosphorylated, this effect can be observed through a microscope. But to see this, the a DEF6 gene has to be mutated and then inserted into COS-7 cells (a specific cell line) that I’m using.
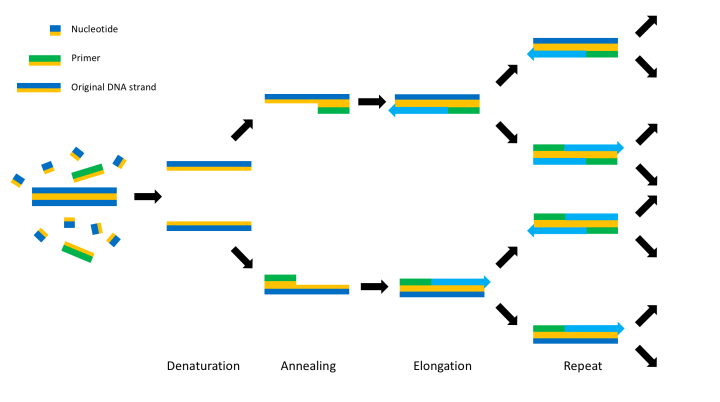
So how does one go about mutating a gene? The actual process is done through a method known as the polymerase chain reaction (or ‘PCR’ to save a lot of hassle) which works by replicating DNA over and over until you have enough to do what you want with it. The double stranded DNA to be replicated is broken down into single strands with its bases free for binding. Small strands of DNA (around 20 bases long) called primers are added to the mix and are specifically designed to be complementary and bind to a site on the DNA. Primers initiate the DNA replication and they are extended using enzymes called polymerases, which add complementary bases to the DNA, building up a new double stranded DNA identical to the original DNA. Because you have two single strands of DNA you end up with two identical double stranded DNA – neat eh? This then means if PCR is performed again then these two DNA strands turn into four which turn into eight and so on.
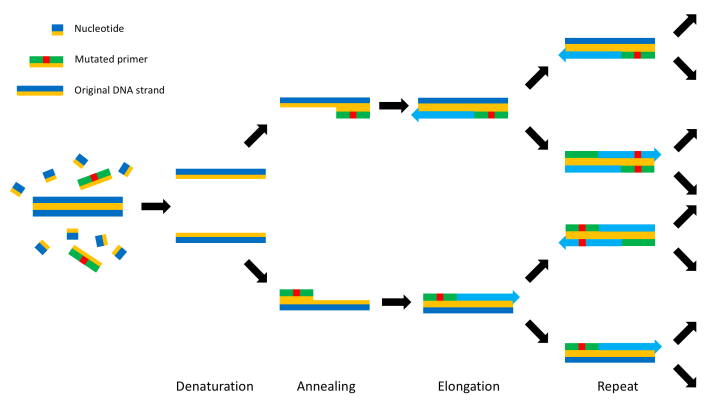
This process can be slightly altered to induce a mutation into the DNA by changing the primer to contain the desired mutation. This will lead the primer to bind either side of the mutation and PCR to carry on as normal but with the mutation fixed in one strand. Successive replications will mean the majority of new strands will contain the mutation – as can be seen below. In this experiment I am using circular bacterial DNA called a plasmid which contains the DEF6 gene, but the method works just the same.
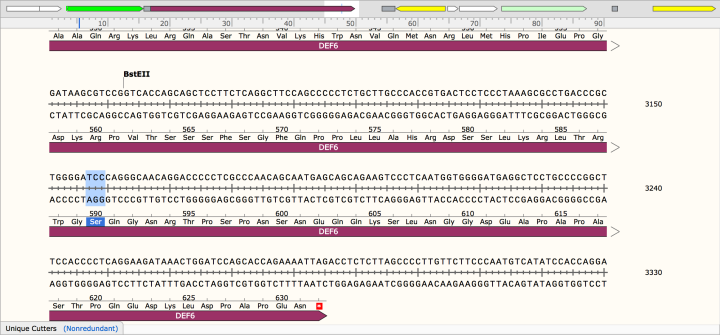
To design the primer that carries the required mutation which will inserted into the DEF6 gene, you first examine the base sequence of the gene in the software SnapGene Viewer (picture below). In SnapGene, the exact positions to insert a mutation to code for a different amino acid can be located. Only positions that have been found to be phosphorylated in DEF6 are being mutated, in the example of the picture below this is serine at position 590 (S590) coded by TCC.
Because the primer needs to bind to either side of the mutation, around three amino acids bases are selected to precede, and four/five amino acids to follow after, this mutation. This also give specificity to the primer so it can only bind at the specific site for mutation. This is a primer I designed earlier for S590, with the serine (TCC) mutated to phenylalanine (TTT):
CGC TGG GGA TTT CAG GGC AAC AGG
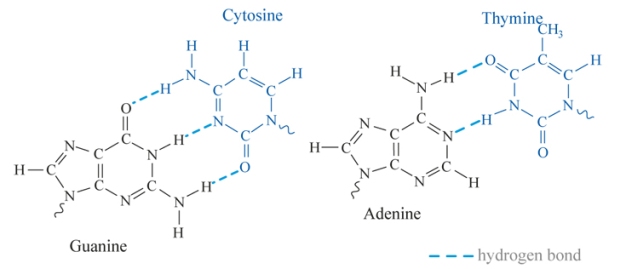
But whoa, hold up there! One can’t just go making primers as simply as that! The guanine/cytosine (G/C) percentage of the primer also needs to be checked. The bases between two DNA strands bond together through interactions called hydrogen bonds, which is where two opposite polar charges attract each other and ‘stick’ together. The more hydrogen bonds present means a higher melting temperature. G/C interactions contain three hydrogen bonds whereas adenine/thymine (A/T) only contain two and therefore the more G/Cs there are, the higher the melting temperature. The G/C percentage should preferably be around 50-70%, for the example primer I’ve used has a G/C content of 62.5%, so we’re all good!
Once the primer is made, PCR can be run until enough plasmid is replicated hopefully with the mutation being fixed into the DNA. From here it is inserted into rapidly dividing bacterial cells in a process known as transformation. These cells are cultured to grow colonies which contain the mutated plasmid. As well as DEF6, the plasmid that is being used in this experiment contains a kanamycin resistance gene (KanR) which gives the bacteria resistance to the antibiotic kanamycin. Growing the bacteria on a kanamycin-containing medium ensures that only transformed bacteria will be able to form colonies. The bacteria are able to replicate the plasmid at a faster rate and unlike PCR, have a lower chance of other mutations arising. A colony of the cultured bacteria is taken with a small portion having the plasmid removed using a lab technique called a mini-prep, which breaks down the cells and separates the plasmid using a centrifuge.
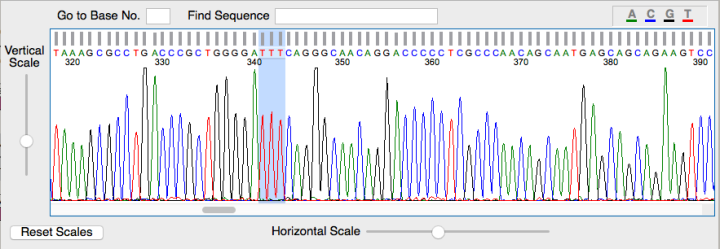
Because PCR isn’t a flawless process and the mutation doesn’t always occur, the extracted plasmids are sent off for sequencing. After the actual scientists have determined the base sequence, it is then checked to see if the mutation is present using software called FinchTV which looks like the picture below. Each coloured peak indicates a particular base and gives the order of the sequence. As you can see in the highlighted area the serine to phenylalanine mutation has occurred.
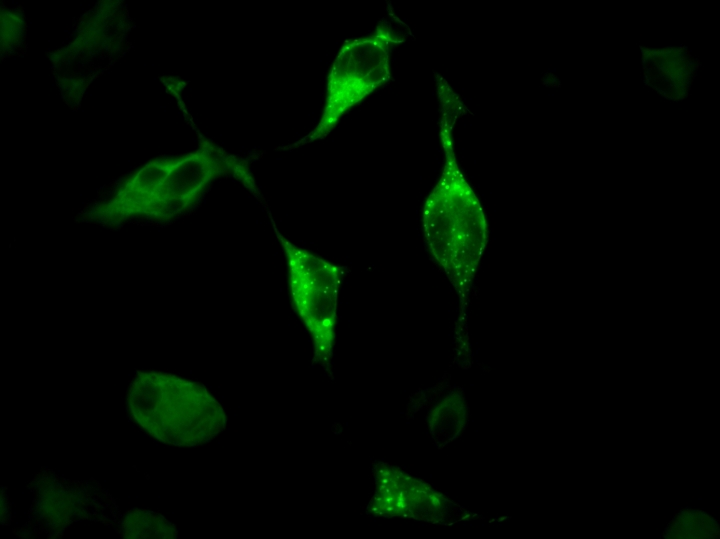
With plasmids definitely containing the mutation needed, they can be inserted into COS-7 cells which resemble to environment DEF6 is naturally in. These transfected cells can be observed with a confocal microscope (a special microscope that lets you see fluorescent proteins) and gives a jazzy image such as one below – that I actually took!
So there you have it, the exciting process of how to make a DEF6 mutant! I realise this is a very niche area but thought you’d like to know on the off chance you’re investigating DEF6 serine/threonine phosphorylations! No? Just me? Damn…
If you’re interested in anything that’s been discussed, here are some links for further reading:
- PCR (although its Wikipedia it has a good summary of the PCR process): https://en.wikipedia.org/wiki/Polymerase_chain_reaction
- DEF6 paper which summarises it in the introduction: http://www.jbc.org/content/287/37/31073.full.pdf
- A simple summary of plasmids and what they can do: https://en.wikipedia.org/wiki/Plasmid
PCR images were made by myself
Base hydrogen bonding image: http://eng.thesaurus.rusnano.com/wiki/article629
SnapGene Viewer and FinchTV pictures taken from the respective software by myself using screen capture
Fluorescent cell picture taken by myself using confocal microscope and the software AxioVision
Title image: http://futurebiomed.com/tag/healthcare-news/
